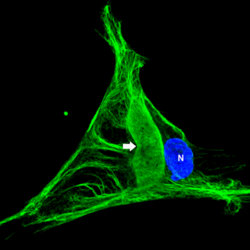
In the fibroblasts derived from the skin biopsies of giant axonal neuropathy (GAN) patients, the vimentin intermediate filaments (green) form large abnormal aggregates (indicated by arrow). In some cases these abnormal aggregates are larger than the nucleus (blue). Presented by postdoctoral fellow Saleemulla Mahammad, PhD, at the American Society for Cell Biology Annual Meeting, new research discusses how the defective protein, gigaxonin, was first identified in children with the rare and untreatable genetic disease known as GAN.
For the first time, a defective protein that plays a specific role in degrading intermediate filaments (IF), one of three classes of filaments that form the structure of nerve cells, has been discovered by an international team of researchers.
Presented by postdoctoral fellow Saleemulla Mahammad, PhD, at the American Society for Cell Biology Annual Meeting, the research discusses how the defective protein, gigaxonin, was first identified in children with a rare and untreatable genetic disease known as giant axonal neuropathy (GAN).
The knowledge of gigaxonin’s specific role explains why a failure in protein degradation would lead to massive aggregations of IF in the neuronal cells of GAN children, said Mahammad, who works in the laboratory of Robert Goldman, PhD, chair of cell and molecular biology.
Mahammad and other members of the Goldman Laboratory collaborated with Puneet Opal, MD, PhD, associate Professor in the Ken and Ruth Davee Department of Neurology and cell and molecular biology, along with researchers in the laboratory of Pascale Bomont at the INSERM neurological institute in Montpelier, France, and the laboratory of Jean-Pierre Julien at the Université Laval in Quebec, Canada.
The GAN gene was first identified in 2000 by the Bomont Laboratory, reporting that it encoded for the protein gigaxonin. Based on sequence homology, gigaxonin is involved in the normal turnover of proteins by the well-studied ubiquitin-proteasome system. But it wasn’t clear why a failure in protein degradation would lead to massive aggregations of IF in a patient’s neuronal cells.
Because it is not possible to study nerve cells experimentally from patients, Mahammad and collaborators instead used fibroblasts from skin biopsies of children with GAN because previous studies have revealed that other classes of IF are also altered in GAN patients. In particular, the IF vimentin expressed in fibroblasts of children with GAN also forms abnormally large aggregates. These cells can readily be obtained from skin biopsies and grown in lab cultures.

When the researchers introduced the gigaxonin gene into both control and patient fibroblasts, the results were dramatic. In the fibroblasts cultured from GAN patients, the complex network of vimentin filaments and abnormal aggregates disappeared. The vimentin filaments in the control cells also disappeared following the overexpression of the gigaxonin protein. Boosting gigaxonin to higher levels in normal cultured nerve cells also led to a degradation of neuronal forms of IF. However, the cytoskeleton’s two other major systems, microtubules and actin filaments, were not affected by this treatment.
These findings point to a central role for gigaxonin in regulating the normal turnover of IF proteins. When gigaxonin is defective, neurofilaments, the specific type of IF located in nerve cells, pile up to form aggregates that eventually disrupts the normal functioning of neurons in GAN.
Gigaxonin is the first factor to be identified that plays a specific role in the degradation of several types of IF proteins, including neurofilaments, according to Mahammad. This discovery may have implications for more common types of neurodegenerative diseases that are also characterized by large accumulations of IF proteins, including Alzheimer’s disease, Parkinson’s disease, dementia with Lewy bodies, Charcot-Marie-Tooth disease, neuronal intermediate filament inclusion disease, and diabetic neuropathy.
GAN is an extremely rare genetic disorder that strikes both the central and peripheral nervous systems of children. The leading GAN disease foundation, Hannah’s Hope Fund, currently knows of 31 cases worldwide, 19 in the United States alone. But its rarity doesn’t dull its severity in children. Although, there are no symptoms at birth, by age three the first signs of muscle weakness usually appear and progress slowly but steadily. With increasing difficulty in walking and coordinating hand movements, children with GAN are often wheelchair-bound by age 10. Over time, they become dependent on feeding and breathing tubes; only a few will survive into young adulthood. The pathological markers for GAN are swollen (thus “giant”) axons, filled with abnormal aggregates rich in neurofilaments.
This research was supported by NIH grant 1P01GM096971-01 and Hannah’s Hope Fund.