Presenting Author:
Kyle Lyman, Ph.D.
Principal Investigator:
Dane Chetkovich, M.D.
Department:
Neurology, Ken and Ruth Davee Department
Keywords:
Depression, Neuroscience, HCN, TRIP8b, Ion Channel
Location:
Third Floor, Feinberg Pavilion, Northwestern Memorial Hospital
B102 - Basic Science
HCN2 mutation reveals essential role for allosterism in TRIP8b binding
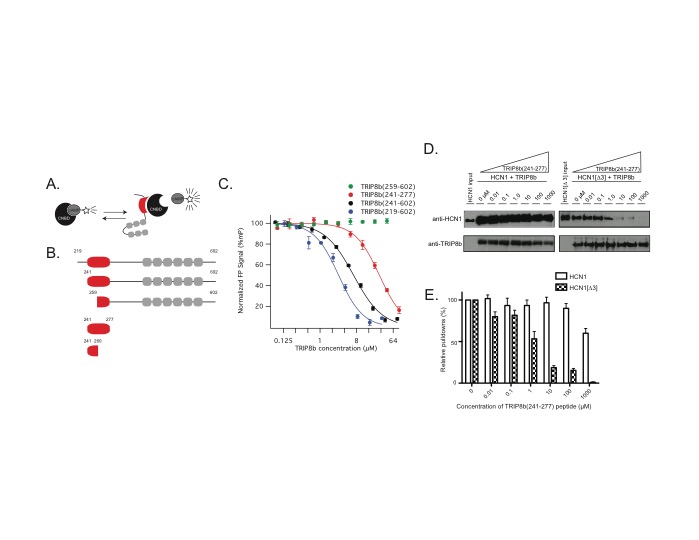
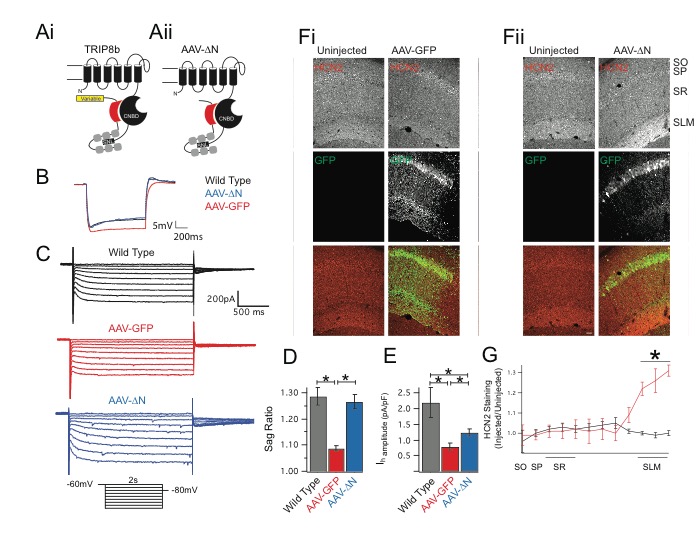
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels regulate cellular excitability and profoundly influence hippocampal CA1 pyramidal neuron function. These noninactivating channels open in response to hyperpolarization and mediate a nonspecific cationic conductance. In addition to their regulation by voltage, channel function is also influenced by the binding of cyclic nucleotides to an intracellular cyclic nucleotide-binding domain (CNBD). Binding of cyclic nucleotides increases the rate of HCN channel activation and depolarizes the half-activation potential of the channel. HCN channels are expressed at higher levels in the distal dendrites and this pattern of expression facilitates the channel’s role in regulating synaptic integration. In order to achieve this distal dendritic enrichment, HCN pore-forming subunits are bound in a 1:1 ratio to an auxiliary subunit, TRIP8b. Recently, the interaction of HCN channels with TRIP8b has been identified as a therapeutic target for the treatment of depression and mice lacking TRIP8b show an increase in antidepressant-like behavior. As such, there is considerable interest in understanding the structure-function relationship of this interaction with the goal of developing new antidepressants. Despite the substantial effort invested in describing the molecular substrates of the TRIP8b-HCN interaction, it remains unclear how precisely this interaction translates into TRIP8b-mediated HCN channel trafficking in CA1 pyramidal neurons. This question is of paramount importance, because decreasing the expression of HCN channels in CA1 distal dendrites increases antidepressant-like behavior while increasing expression has the opposite effect. Initial reports suggested that the variably spliced N terminus of TRIP8b was the primary determinant of subcellular trafficking, with different exons allowing for the interaction with novel binding proteins that would facilitate channel transport. However, in a previous report our group noted that disruption of either TRIP8b-HCN binding site was sufficient to limit trafficking of HCN channels. This raised the possibility that there is a structural requirement for trafficking which is independent of the variably spliced region. In this report, we establish that the N terminus of TRIP8b is not required for distal dendritic enrichment or surface trafficking of HCN channels in CA1 pyramidal neurons. Using a novel fluorescence polarization based assay, we then identify an intramolecular conformational change in TRIP8b that allosterically influences HCN channel binding. We then use a novel knock-in mouse to disrupt the TRIP8b conformational change, and show that this is sufficient to impair trafficking of HCN channels into the distal dendrites of CA1 pyramidal neurons. These results have important consequences for the development of novel antidepressants, as they suggest that disrupting either TRIP8b-HCN binding site will be sufficient to produce symptomatic relief.